Introduction
Schwannoma (or neurilemmoma) is a benign tumor that is comprised of cells with the morphology and phenotype of the Schwann cell [1]. Cytogenetic and comparative genomic hybridization studies of adult schwannomas, either classic or cellular, indicate a complete or partial loss of chromosome 22, which bears the tumor suppressor gene NF2 as the most frequent abnormality [2]. In this study, we report a case of a giant invasive sacral schwannoma with a cytogenetically rare occurrence of trisomy 22.
Case Report
A 58-year-old woman had a 6-year history of progressive left buttock pain radiating to the limb. The pain was aggravated by walking, while relieved by lumbar flexion. She had no history of antecedent trauma or constitutional symptoms. She had objective weakness of the bilateral extensor hallucis longs, graded as 4/5 by manual muscle testing. Sensory deficit was observed in the left S1-3 area. Deep tendon reflexes were preserved and were normal bilaterally at the knees and ankles. The patient had no fasciculation, atrophy, or upper motor neuron signs. No cutaneous stigmata of neurofibromatosis were observed.
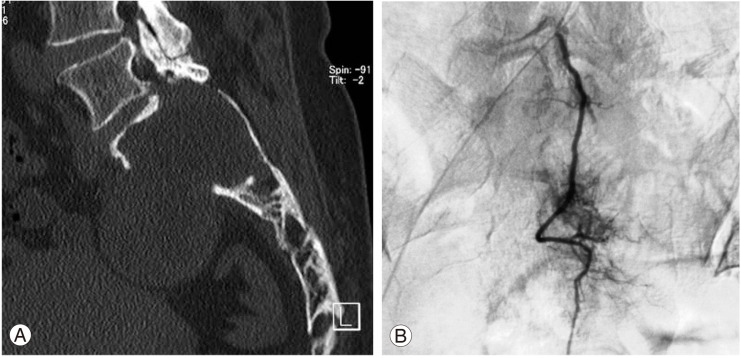
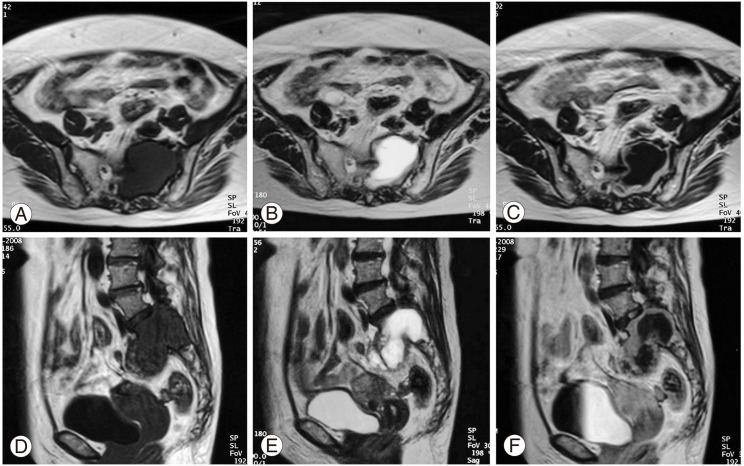
In the computed tomography (CT) scans, a large mass was found in the upper sacrum (Fig. 1A). Angiogram showed evidence of hypervascularity (Fig. 1B). Magnetic resonance imaging (MRI) revealed a tumorous cystic lesion (9.5×8.0×4.5 cm) occupying most of the left upper sacrum. The cystic wall showed a hypointense signal on the T1-weighted images and a moderately high signal on the T2-weighted images. Homogeneous rim enhancement of the cystic lesion was observed following administration of gadolinium contrast (Fig. 2). A tru-cut bone biopsy determined a histological diagnosis of schwannoma.
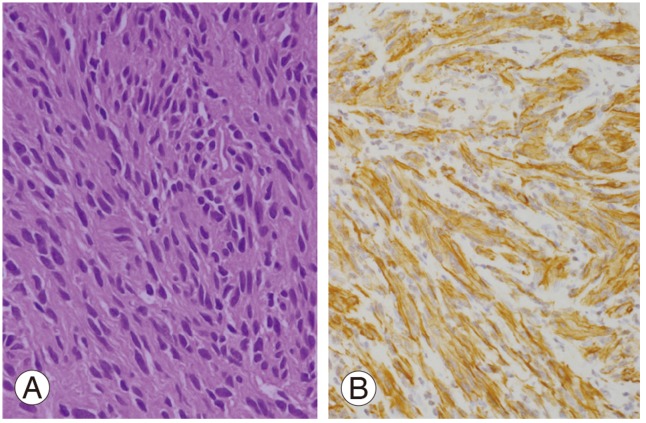
Bilateral laminectomy (L4-S3) was performed using a posterior midline approach to expose the spinal canal. The cystic tumor was found to be firmly adherent to the arachnoid and nerve roots. Histopathological examination showed an Antoni A schwannoma (hypercellular) in most areas. The tumor cells were richly cellular but with a low mitotic count (Fig. 3A). Smaller areas showed the presence of an Antoni B schwannoma (hypocellular). No anaplasia or necrosis was detected. Immunohistochemical staining showed strong expression of the S-100 protein and glial fibrillary acidic protein (GFAP) (Fig. 3B). There was no evidence of nerve infiltration or mitoses to suggest malignancy. The tumor was confirmed as a giant invasive spinal schwannoma (GISS) composed of spindle cells with elongated, hyperchromatic nuclei and abundant cytoplasm.
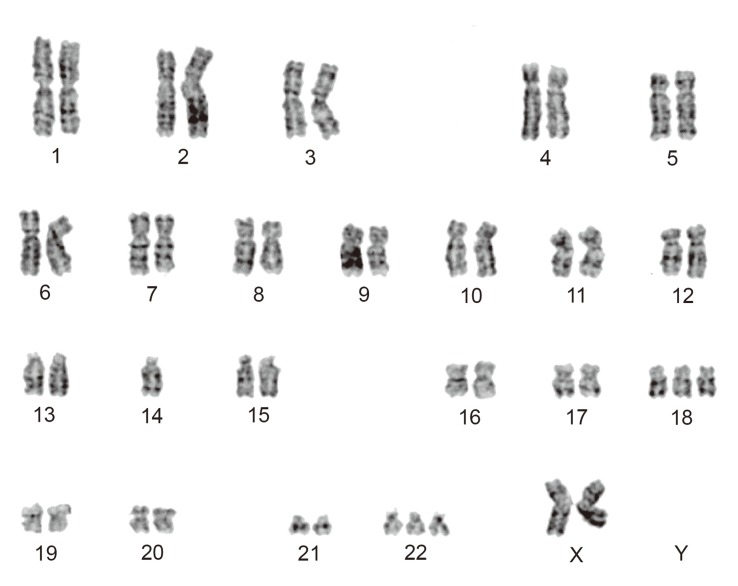
Cytogenetic analyses were performed on primary cultures obtained from the first surgical sample. Standard culture and harvesting procedures described previously were utilized [2]. Karyotype designation was established in accordance with the International System for Human Cytogenetic Nomenclature 2009 [3]. Twenty cells were analyzed of which 8 cells represented an abnormal clone characterized by a chromosomal numerical aberration, 47,XX,-14,+18,+22 (Fig. 4). Ten cells showed various abnormal patterns, which were derived from -14, +18, or +22, where the remaining 2 cells were found to be normal female cells (46,XX).
Although the tumor was incompletely excised, the patient was generally in good condition without disease progression for 3.3 years after surgery.
Discussion
Schwannoma is a benign tumor that arises from the embryonic neural crest cells of the nerve sheaths of peripheral or cranial nerves. In some cases, it presents as a huge mass that extends into the vertebral body and extraspinal space, as has been described in several case reports [4-7]. According to the definition provided by Sridhar et al. [4], GISS extends over more than two vertebral levels, erodes vertebral bodies, and extends posteriorly and laterally into the myofascial planes. Our case involved the sacrum, and may thus differ from typical spinal schwannomas.
Schwannoma in its classic form consists of spindle-shaped cells. The histological hallmark of the schwannoma is the presence of alternating areas of Antoni A (compact, hypercellular, well-organized spindle cells in a palisading pattern), palisading nuclei (Verocay bodies), and Antoni B (hypocellular, loose-textured pleomorphic cells with predominantly myxoid cytoplasm). Immunohistochemical staining showed diffuse immunoreactivity for the S-100 protein and GFAP [1]. However, many histological variants of schwannomas have been described, such as ancient schwannoma, cellular schwannoma, plexiform neurinoma, melanocytic neurinoma. Cystic changes in an ancient schwannoma is likely attributable to mucinous degeneration, ischemic necrosis, hemorrhage, and microcystic formation. However, the cellular schwannoma observed in this case study differed from the classic type in that the dense Antoni A pattern comprised ≥90% of the tumor area with a more uniform pattern and a lack of Verocay bodies [7]. In our case, histologically, the tumor was a cellular schwannoma with marked cystic changes. Complete surgical excision was recommended to prevent recurrence in such cases, but this was not feasible without sacrificing nerve roots over most of the sacral region.
Monosomy 22 has been reported previously, providing cytogenetic evidence in support of schwannomas. Loss of chromosomes 4, 6, 10, 12, 15, 17, 18, X and Y was also observed in all cases described in the literature [2,8-10]. On the other hand, trisomy 5, 7, 13, and 20 has also been reported. The findings of this study suggests a possible association of schwannomas with trisomy 22, which is important because NF2 has been mapped on22q11.
In conclusion, trisomy 22 as a cytogenetic abnormality seems to be specifically associated with cellular schwannoma as well as monosomy 22. This association needs to be confirmed by cytogenetic analysis of further cases of this tumor, which could contribute to the correct nosological classification of this entity and would provide a better understanding of its pathogenesis.