Transforming Growth Factor-β Induces Interleukin-6 Secretion from Human Ligamentum Flavum–Derived Cells through Partial Activation of p38 and p44/42 Mitogen-Activated Protein Kinases
Article information

Abstract
Study Design
This experimental study was performed using human ligamentum flavum–derived cells (HFCs).
Purpose
To investigate the intracellular signaling mechanism of interleukin-6 (IL-6) secretion in transforming growth factor-β (TGF-β)-stimulated HFCs.
Overview of Literature
Lumbar spinal stenosis (LSS) is a prevalent disease among the elderly, characterized by debilitating pain in the lower extremities. Although the number of patients with LSS has increased in recent years, the underlying pathomechanism remains unclear. Clinical examinations typically rely on magnetic resonance imaging to diagnose patients, revealing ligamentum flavum hypertrophy. Some studies have suggested an association between ligamentum flavum hypertrophy and inflammation/fibrosis, and expression of TGF-β and IL-6 has been observed in surgically obtained ligamentum flavum samples. However, direct evidence linking TGF-β and IL-6 expression in HFCs is lacking.
Methods
HFCs were obtained from patients with LSS who had undergone decompression surgery. The cells were stimulated with TGF-β and pretreated with either the p38 mitogen-activated protein (MAP) kinase inhibitor SB203580 or the p44/42 MAP kinase inhibitor FR180204. IL-6 secretion in the cell culture medium and IL-6 messenger RNA (mRNA) expression levels were analyzed using an enzyme-linked immunoassay and real-time polymerase chain reaction, respectively.
Results
TGF-β administration resulted in a dose- and time-dependent stimulation of IL-6 release. Treatment with SB203580 and FR180204 markedly suppressed TGF-β–induced IL-6 secretion from HFCs. Moreover, these inhibitors suppressed IL-6 mRNA expression in response to TGF-β stimulation.
Conclusions
Our findings indicate that TGF-β induces IL-6 protein secretion and gene expression in HFCs through the activation of p38 or p44/42 MAP kinases. These results suggest a potential association between IL-6–mediated inflammatory response and tissue hypertrophy in LSS, and we provide insights into molecular targets for therapeutic interventions targeting LSS-related inflammation through our analysis of the MAP kinase pathway using HFCs.
Introduction
The global population is rapidly increasing, with an increase in the number of elderly people leading to a rise in the prevalence of lumbar spinal stenosis (LSS), a debilitating condition that severely affects the daily activities and quality of life of affected individuals [1]. LSS primarily manifests as lower back pain and lower extremity pain, often accompanied by gait disturbances, such as intermittent claudication. Diagnosis of LSS relies heavily on magnetic resonance imaging, which reveals lumbar spinal canal stenosis resulting from ligamentum flavum (LF) hypertrophy, leading to compression of the spinal cord or cauda equina [2,3]. Although inflammatory responses, including those related to interleukin-6 (IL-6), are known to be important pain mediators in sciatic symptoms [4], and specific features, such as LF hypertrophy, are common in patients with LSS, the precise relationship between LF hypertrophy and pain in these patients remains unclear. Standard treatments for LSS primarily focus on pain reduction through nonsteroidal anti-inflammatory drugs, gabapentinoids, and certain opioids [5]. Surgical interventions, such as decompression, are considered when conservative approaches fail. To develop novel therapeutic strategies for LSS, a comprehensive understanding of the mechanisms underlying LF hypertrophy is crucial.
Some studies have linked LF hypertrophy to inflammation and fibrosis, as indicated by the expression of inflammation- and fibrosis-related genes in LF tissues [6]. Histological studies in elderly patients with LSS have shown a decrease in LF elasticity and an increase in collagenous fibers [7,8]. In studies on heart failure, tissue injury and scar formation have also been investigated in relation to myocardial infarction [9]; transforming growth factor-β (TGF-β) was implicated in scar formation following the initial inflammatory response. However, in LF hypertrophy, TGF-β has primarily been associated with early-stage processes rather than scar formation [10]. These observations suggest that injury, scar formation, and LF hypertrophy may be closely associated. Previous molecular studies have explored LF cells, demonstrating that TGF-β stimulates collagen messenger RNA (mRNA) expression in LF-derived fibroblasts, suggesting a connection to scar formation [11].
To develop targeted therapeutic approaches, it is essential to investigate the molecular mechanisms underlying the disease. For instance, p38 and p44/42 mitogen-activated protein (MAP) kinases have been shown to get activated by recombinant human growth and differential factor in LF cells [12]. TGF-β has also been found to stimulate connective tissue growth factors in human ligamentum flavum–derived cells (HFCs) through p38 MAP kinase activation [13]. However, no studies have examined the intracellular signaling involved in TGF-β–induced IL-6 secretion in the LF.
The present study aimed to elucidate the intracellular signaling mechanism underlying TGF-β–induced IL-6 secretion in HFCs. The specific focus was on intracellular mechanisms involving p38 and p44/42 MAP kinases.
Materials and Methods
1. Study design
This study was conducted in accordance with the principles outlined in the Declaration of Helsinki. The study protocol received approval from the Institutional Review Board of Nagoya City University (IRB code: 60-18-0215). LF tissues were obtained from patients who underwent surgery for LSS, and each patient provided written informed consent. Samples were obtained from nine patients (four men and five women; mean age, 75 years). The mean thickness of the LF, as measured using magnetic resonance imaging, was 5.2 mm.
2. Cell culture of human LF-derived cells
HFCs were isolated following an established protocol [14,15]. The excised tissue was carefully trimmed, and any blood was washed away using phosphate-buffered saline (PBS). Ossified or calcified tissues were removed. The tissue was then cut into approximately 3 mm3 pieces using scissors. Digestion of the tissue was performed using 0.2% collagenase type I in Dulbecco’s modified Eagle medium (DMEM) for 1 hour. Subsequently, the tissue was washed with PBS, and a second digestion step was performed using 0.025% collagenase type I in DMEM supplemented with 5% fetal bovine serum (FBS; Gibco, Gaithersburg, MD, USA) overnight. The medium was then filtered through a 70 μm membrane, and the cells were cultured in a 75 cm2 flask containing DMEM supplemented with 10% FBS. Cells from passages 2–5 were used for all experiments. Collagenase type I, PBS, and DMEM were purchased from Wako Pure Chemical Industries (Osaka, Japan).
3. Stimulation of HFCs with TGF-β
HFCs were seeded in 6-well plates at a density of 2.0×105 cells/well in 10% FBS–DMEM. When sub-confluency was reached, the culture medium was replaced with 0.3% FBS–DMEM. For TGF-β (Funakoshi, Tokyo, Japan) stimulation experiments, the p38 MAP kinase inhibitor SB203580 (10 μM) or the p44/42 MAP kinase inhibitor FR180204 (Cayman Chemical, Ann Arbor, MI, USA) was added 1 hour prior to the addition of TGF-β at concentrations of 0.01–10 ng/mL.
4. Western blot analysis
Cells were harvested using radioimmunoprecipitation assay buffer (NACALAI, Kyoto, Japan) supplemented with a protease inhibitor. Sodium dodecyl sulfate–polyacrylamide gel electrophoresis was conducted using 10% polyacrylamide gels, followed by Western blot analysis involving primary antibodies with peroxidase-labeled secondary antibodies. Primary antibodies against phospho-p44/42 MAP kinase, phospho-p38 MAP kinase, p44/42 MAP kinase, and p38 MAP kinase, as well as secondary antibodies, were purchased from Cell Signaling (Danvers, MA, USA). Peroxidase activity on the polyvinylidene fluoride membrane was detected using ECL substrate (Bio-Rad, Hercules, CA, USA). Images were visualized using an Amersham Imager 680 blot and gel imager (Cytiva, Tokyo, Japan). Band density analysis was conducted using ImageJ (NIH; https://imagej.nih.gov/ij/download.html).
5. IL-6 assay
The concentration of IL-6 in the conditioned medium secreted by HFCs was determined using a human IL-6 enzyme-linked immunosorbent assay kit (D6050; R&D, Minneapolis, MA, USA).
6. Gene expression analysis
Cultured cells were collected using TRIzol reagent (1 mL), chloroform (0.2 mL) was added to the Trizol, and the samples were centrifuged. The aqueous phase containing total RNA was obtained and purified using the PureLink RNA Mini Kit (Thermo Fisher Scientific, Tokyo, Japan). Complementary DNA was synthesized using iScript RT Supermix for real-time quantitative polymerase chain reaction (RT-qPCR) (Bio-Rad), and specific primers (GAPDH; Hs.PT.39a.22214836, IL-6; Hs.PT.58.40226675, IDT) were used for amplification with Fast SYBR Green Master Mix. RT-qPCR was performed using a QuantStudio 12 K Flex Real-Time PCR System (Thermo Fisher Scientific, Tokyo, Japan). The PCR conditions were as follows: one cycle at 95°C for 20 seconds, followed by 40 cycles at 95°C for 1 second and 60°C for 20 seconds. The relative mRNA expression levels of IL-6 were normalized to GAPDH mRNA levels and calculated using the 2-ΔΔCt method. TRIzol, PureLink RNA Mini Kits, and Fast SYBR Green Master Mix were purchased from Thermo Fisher Scientific.
7. Quantitative data presentation and statistical analyses
Data are presented as the mean±standard deviation from biological triplicates. Statistical analysis was performed using a two-tailed Student t-test, and a p>0.05 was considered significant. The experiments were repeated at least twice using different cell preparations to ensure reproducibility.
Results
1. TGF-β–stimulated IL-6 secretion in HFCs
First, we investigated the effect of TGF-β administration on IL-6 secretion in HFCs. TGF-β (10 ng/mL) was added to the culture medium, and samples were collected up to 48 hours after stimulation. IL-6 secretion was time-dependent, with TGF-β inducing secretion for up to 48 hours (Fig. 1A). Furthermore, we assessed the dose-dependent effect of TGF-β concentration on IL-6 secretion at the 48-hour time-point. TGF-β stimulated IL-6 secretion in HFCs in a dose-dependent manner (0.01–10 ng/mL) (Fig. 1B).

Transforming growth factor-β (TGF-β) stimulates interleukin-6 (IL-6) secretion from human ligamentum flavum–derived cells (HFCs). (A) TGF-β (10 ng/mL) was added to the culture medium, after which the conditioned medium was collected at the indicated time, and IL-6 level was measured by enzyme linked immunosorbent assay. TGF-β (10 ng/mL) stimulates IL-6 secretion up to 48 hours after TGF-β stimulation. (B) Various doses of TGF-β were added to HFCs, and the conditioned medium was collected at the 48-hour time point. TGF-β stimulated IL-6 secretion in a dose-dependent manner.
2. TGF-β–induced phosphorylation of p38 and p44/42 MAP kinases in HFCs
To investigate the involvement of MAP kinase activation during TGF-β stimulation in HFCs, we analyzed the phosphorylation of p38 and p44/42 MAP kinases. In the absence of TGF-β stimulation, p38 MAP kinase phosphorylation was not detected. However, upon addition of TGF-β, phosphorylation of p38 MAP kinase increased in a time-dependent manner (Fig. 2A), reaching its peak at 2–3 hours after TGF-β stimulation. Similarly, TGF-β administration stimulated the phosphorylation of p44/42 MAP kinase in a time-dependent manner (Fig. 2B), with the maximum phosphorylation observed 2 minutes after TGF-β stimulation.

Transforming growth factor-β (TGF-β) induces the phosphorylation of p38 mitogen-activated protein (MAP) and p44/42 MAP kinases. Phosphorylation of p38 and p44/42 MAP kinases was detected by western blotting. TGF-β (10 ng/mL) time-dependently induced the phosphorylation of p38 MAP kinase (p-p38) (A) and the phosphorylation of p44/42 MAP kinase (p-p44/42) (B). The results are expressed as the mean±standard deviation of three independent experiments.
3. Involvement of p38 and p44/42 MAP kinases in IL-6 secretion from HFCs
To investigate the role of p38 MAP kinase and p44/42 MAP kinase in IL-6 secretion from TGF-β–stimulated HFCs, specific inhibitors of these MAP kinases were used. Prior to TGF-β (10 ng/mL) stimulation, SB203580 (10 μM) or FR180204 (10 μM) was added to the culture medium for 1 hour. After 48 hours of TGF-β stimulation, the conditioned medium was collected. Although SB203580 (10 μM) or FR180204 (10 μM) alone did not affect IL-6 secretion from HFCs, they significantly reduced TGF-β–stimulated IL-6 secretion (Fig. 3).
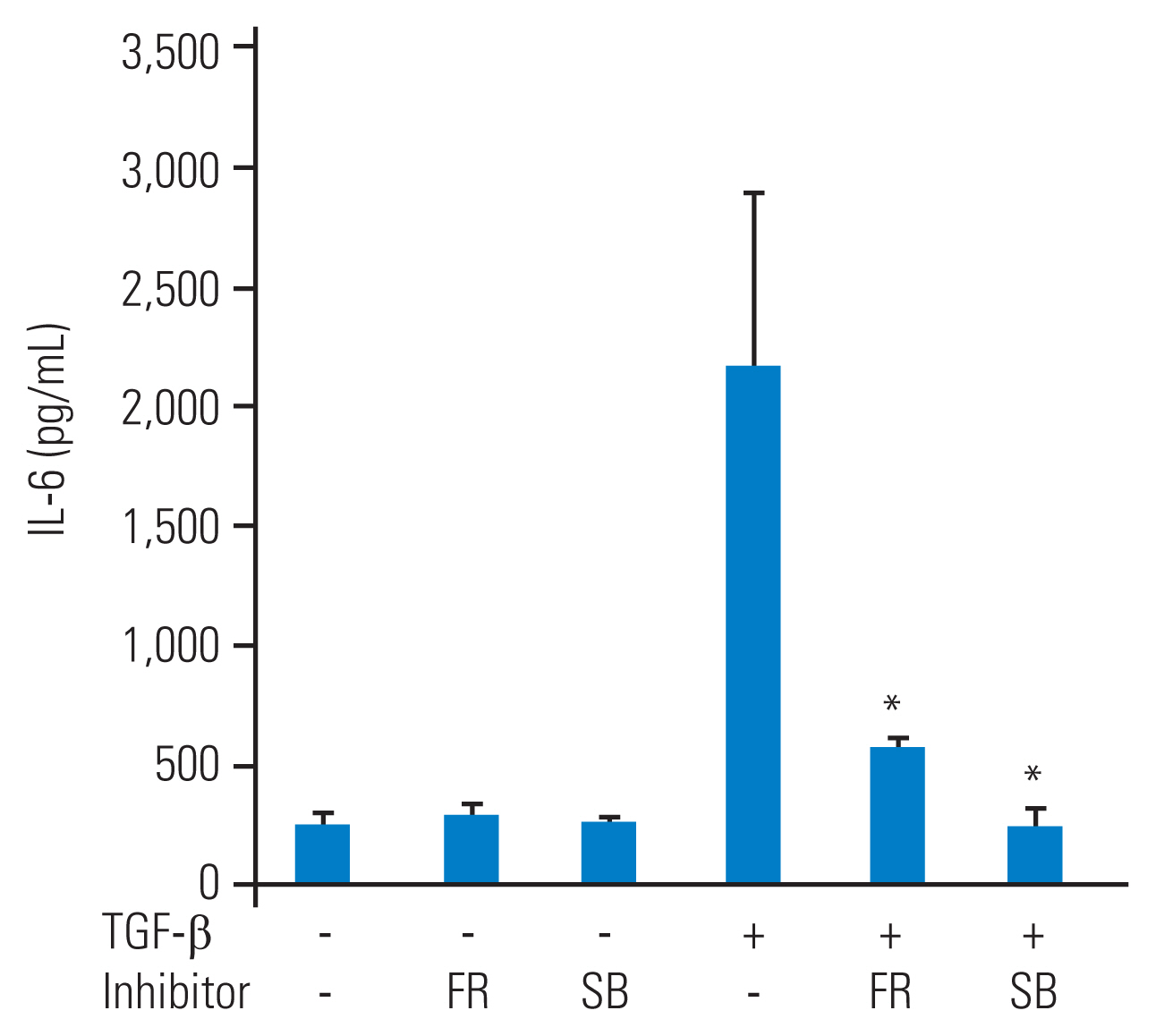
p38 mitogen-activated protein (MAP) kinase or p44/42 MAP kinase regulate transforming growth factor-β (TGF-β) stimulated interleukin-6 (IL-6) secretion from human ligamentum flavum–derived cells (HFCs). SB203580, a p38 MAP kinase inhibitor, or FR180204, a p44/42 MAP kinase inhibitor, was supplied into the medium and TGF-β (10 ng/mL) was added 1 hour later. The conditioned medium was measured at the 48-hour time point. SB203580 (SB) or FR180204 (FR) significantly suppressed the IL-6 secretion from HFCs stimulated by TGF-β. *p<0.05, to TGF-β alone.
4. Regulation of TGF-β–stimulated IL-6 mRNA expression by p38 and p44/42 MAP kinases
To determine the extent to which p38 and p44/42 MAP kinases regulated TGF-β–stimulated IL-6 production in HFCs, we tested the hypothesis that they regulate IL-6 gene expression, given that the MAP kinases are activated relatively early following TGF-β stimulation. Total RNA was extracted 9 hours after TGF-β stimulation, and gene expression was analyzed using RT-qPCR. TGF-β effectively stimulated IL-6 gene expression in HFCs, which was significantly suppressed by SB203580 and FR180204 (Fig. 4).

Transforming growth factor-β (TGF-β)-stimulated interleukin-6 (IL-6) gene expression is regulated by p38 mitogen-activated protein (MAP) kinase or p44/42 MAP. SB203580, a p38 MAP kinase inhibitor, or FR180204, a p44/42 MAP kinase inhibitor, was added to HFCs as a pretreatment, and TGF-β (10 ng/mL) was added 1 hour later. Cells were harvested by TRIzol after 9 hours of TGF-β stimulation. Gene expression was measured by real-time polymerase chain reaction. Normalized gene expression is shown as 2-ΔΔCt (GAPDH was used as a housekeeping gene). TGF-β stimulated IL-6 messenger RNA (mRNA) expression. SB203580 (SB) or FR180204 (FR) significantly suppressed IL-6 mRNA level stimulated by TGF-β. *p<0.05, to TGF-β alone.
Discussion
In the present study, we demonstrated the involvement of p38 and p44/42 MAP kinases in both gene expression and secretion of IL-6 in HFCs upon TGF-β stimulation. The activation of p44/42 MAP kinase occurred within minutes after TGF-β stimulation, whereas the activation of p38 MAP kinase persisted for a longer duration of approximately 2–3 hours. This discrepancy in activation kinetics may be attributed to the direct phosphorylation signaling that activates p44/42 MAP kinase, whereas p38 MAP kinase activation involves the induction of specific proteins.
LF hypertrophy is believed to be associated with injury and scar formation, as evidenced by histological changes characterized by alterations in the ratio of collagen fibers to elastic fibers. In hypertrophied LF tissue, there is a significant increase in collagen fibers, with collagen-related gene expression also found to be significantly higher in the dorsal layer [16]. During scar formation, TGF-β plays an important role in many tissues, including the skin, tendons, and heart [9,17,18], and the expression of TGF-β has been reported in the LF [7,8,11]. Moreover, microarray gene analysis of adolescent idiopathic scoliosis surgical samples has revealed that the gene expression levels of inflammatory cytokines, including TGF-β and IL-6, are increased in hypertrophied LF [19].
Attempts have been made to develop animal models of LSS [20–22]. Such models involve direct narrowing of the spinal canal and are used to study the spinal cord. However, they cannot be used to investigate the LF, which is a crucial component in LSS management. Thus, there is a need for an animal model that specifically induces LF hypertrophy. However, establishing animal models of LSS is challenging owing to the significant differences in spine loading between humans and animals. To overcome this limitation, a bipedal standing mouse model was tested in a previous study [23] and found to exhibit higher gene expression levels of inflammatory cytokines and fibrosis-related factors, including interleukins and collagens. Another study demonstrated that mechanical loading stress in mice led to LF hypertrophy [24] and increased the expression levels of various genes, such as TGF-β and IL-6 genes, suggesting the importance of macrophage infiltration and inflammatory responses in LF hypertrophic changes.
Although these animal models provide molecular insights into LF hypertrophy, there have been no previous reports directly correlating TGF-β and IL-6. In the present study, to the best of our knowledge, we show for the first time that TGF-β stimulates IL-6 secretion in HFCs. However, our results regarding the levels of TGF-β–stimulated IL-6 were not consistent (Figs. 1, 3). This inconsistency may be attributed to the use of primary HFCs isolated from surgical samples, as opposed to established cell lines, leading to variations in cell activity among samples. To address this, we conducted repeated experiments to ensure reproducibility. Our findings suggest that TGF-β, which is upregulated during injury and scar formation, can induce IL-6 production in the LF, and in turn, IL-6 can contribute to the inflammatory response, ultimately resulting in LF hypertrophy. Notably, IL-6 is an important pain mediator associated with sciatic nerve symptoms [4]. Increased levels of IL-6 protein have been observed in the facet joints or cerebrospinal fluid of patients with LSS [25,26], and high expression levels of IL-6 have been detected in the LF of these patients [6]. Additionally, a study involving a rat model showed a correlation between IL-6 levels and lower extremity pain, as assessed through animal behavior tests [27]. Although it is essential to treat LF hypertrophy, pain reduction is a primary concern for patients. Therefore, regulating TGF-β–stimulated IL-6 secretion from the LF could be beneficial not only for preventing hypertrophy but also for pain management.
There is a growing interest in understanding the intracellular signaling mechanisms to facilitate the development of therapeutic strategies in medicine. MAP kinases, including p38 and p44/42 MAP kinases, have been found to be activated by certain agents in HFCs [12,13]. Zhong et al. [12] demonstrated the involvement of p38 and p44/42 MAP kinases in the osteogenic differentiation of HFCs induced by growth/differentiation factor (GDF)-5. They found that GDF-5 stimulation led to increased alkaline phosphate activity, matrix mineralization, and osteocalcin expression through activation of p38 and p44/42 MAP kinases. Cao et al. [13] reported the participation of p38 MAP kinase in TGF-β–induced expression of connective tissue growth factor, collagen I, and collagen III, which are associated with LF hypertrophy, in HFCs. Ito et al. [28] suggested a link between oxidative stress and LF hypertrophy, where the phosphorylation of p38 and p44/42 MAP kinases played a role. Zheng et al. [29] proposed a potential mechanism of LF hypertrophy, demonstrating the regulation of profibrotic proteins through the activation of p44/42 MAP kinase by cytokine receptor-like factor I. Consistent with these findings, our study reveals the involvement of p38 and p44/42 MAP kinases in TGF-β–stimulated IL-6 secretion in HFCs. Importantly, the recent interest in MAP kinases in the context of LF highlights the potential therapeutic targeting of MAP kinase activity for LSS treatment.
Conclusions
HFCs were used to investigate the intracellular signaling involved in TGF-β–induced IL-6 secretion. The regulatory role of p38 and p44/42 MAP kinases in the mRNA and protein expression of IL-6 in TGF-β–stimulated HFCs was examined.
Acknowledgments
This work was supported by the Japan Society for the Promotion of Science Grants-in-Aid for Scientific Research–KAKENHI (grant number: 17K16697 and 19K18506). The authors are grateful to acknowledge the assistance of the Research Equipment Sharing Center at the Nagoya City University.
Notes
Conflict of interest
No potential conflict of interest relevant to this article was reported.
Author Contributions
Yuta Goto and Kenji Kato wrote and prepared the manuscript. Yuta Goto and Kenji Kato planned, collected, and analyzed experimental data. Yuta Goto, Kenji Kato, Kiyoshi Yagi, Yohei Kawaguchi, Hiroki Yonezu, Tomoko Koshimae, Yuko Waguri-Nagaya, Hideki Murakami, and Nobuyuki Suzuki discussed the design of this study. All authors have read, reviewed, and approved the article.