Introduction
Tuberculosis remains in the list of the diseases, causing untold human suffering even in the era of remarkably advanced medical science, and the reasons for this remain to be properly addressed. The fact is that clinicians and medical scientists as yet have not been able to win the war against the tubercle bacilli, the causative agent of tuberculosis. As regards to this issue, based on the authors' own clinical and research experiences, they have revisited all the available literatures to address the issues in curing and preventing tuberculosis and to provide the most recent information about tubercle bacilli and its role in causing tuberculosis.
Tuberculosis as a disease was once known as the ŌĆ£consumption,ŌĆØ emaciation disease, as the disease victims became cachectic, displaying remarkable weight loss. At that time, the agent causing emaciation was not known [12345678]. It is now known that tubercle bacilli, a bacterium (Mycobacterium tuberculosis, M. tuberculosis), under normal oxygen tension have the ability to shift into one of two non-replicating stages: microaerophilic and anaerobic persistence. From these two forms, the anaerobic persistence is responsible for the mycobacteria's ability to lie dormant in the host for long periods of time, with the capacity to revive and activate the disease at a later time [5679].
Tubercle bacilli are obligate aerobes that can become facultative anaerobes. In terms of different sites for the bacillus to reside, bacillary behavioral characteristic in pulmonary tuberculosis are not different from osteoarticular tuberculosis. However, the bacillary activities in the two organs, as lesions, are somewhat different because of the different environments such as oxygen tension. Bacilli are rather less active in the osteoarticular lesions than the pulmonary ones. Thus, bacillary populations in osteoarticular lesions are less (paucibacillary) than that of the pulmonary ones [79101112].
When the human body becomes infected with tubercle bacilli, the war between the host phagocytes (as defenders) and the bacilli (as invaders) is waging. At this stage, the host phagocytes are reinforced by their chemical weapons, namely lytic digestive enzymes and free oxygen radical providing enzymes. In the case of bacillary lung exposure, the first phagocytic alveolar macrophages recognize bacilli, and engulf and in almost 90% of cases, kill them as part of the non-specific host defense.
Tubercle bacilli also use chemical weapons to defend, survive, grow and thrive, but are slow in action. On the other hand, the host defenses with phagocytic and humoral immune systems are more rapidly mobilized in an attempt to catch and kill or expel the mycobacterial pathogens. The final outcome could be the host or the pathogen as both the defenders (host) and the offenders (bacilli) have their own physical and chemical weapons for their own defense and to disarm the opponent's weapons, in order to win the war.
Tuberculosis is a tissue pathology, produced by the ŌĆ£tug-of-warŌĆØ between the host phagocytes and the inseminated M. tuberculosis. In the war between the host phagocytes and tubercle bacilli, there are three barriers used by the host: physical, chemical and cellular barriers. These barriers are designed to prevent a pathogen's access to host tissue. In particular, the chemical barriers in host are the tissue pH (as in stomach), proteolytic enzymes (as in eye tear), generated superoxide, and antimicrobial cationic (positively charged) peptides called defensins (a component of innate immunity) against mycobacterial infection.
In the war between phagocytes and mycobacteria, host defense is essential in protecting the host body, and it defines the success or failure for the host. Tubercle bacilli have also evolved ways to adapt and survive in the host environment with effects such as developing drug resistance, simply through increased production of mycobacterial sulfatides, cord factor, and chaperonins 10 [13141516].
Morphology of M. tuberculosis
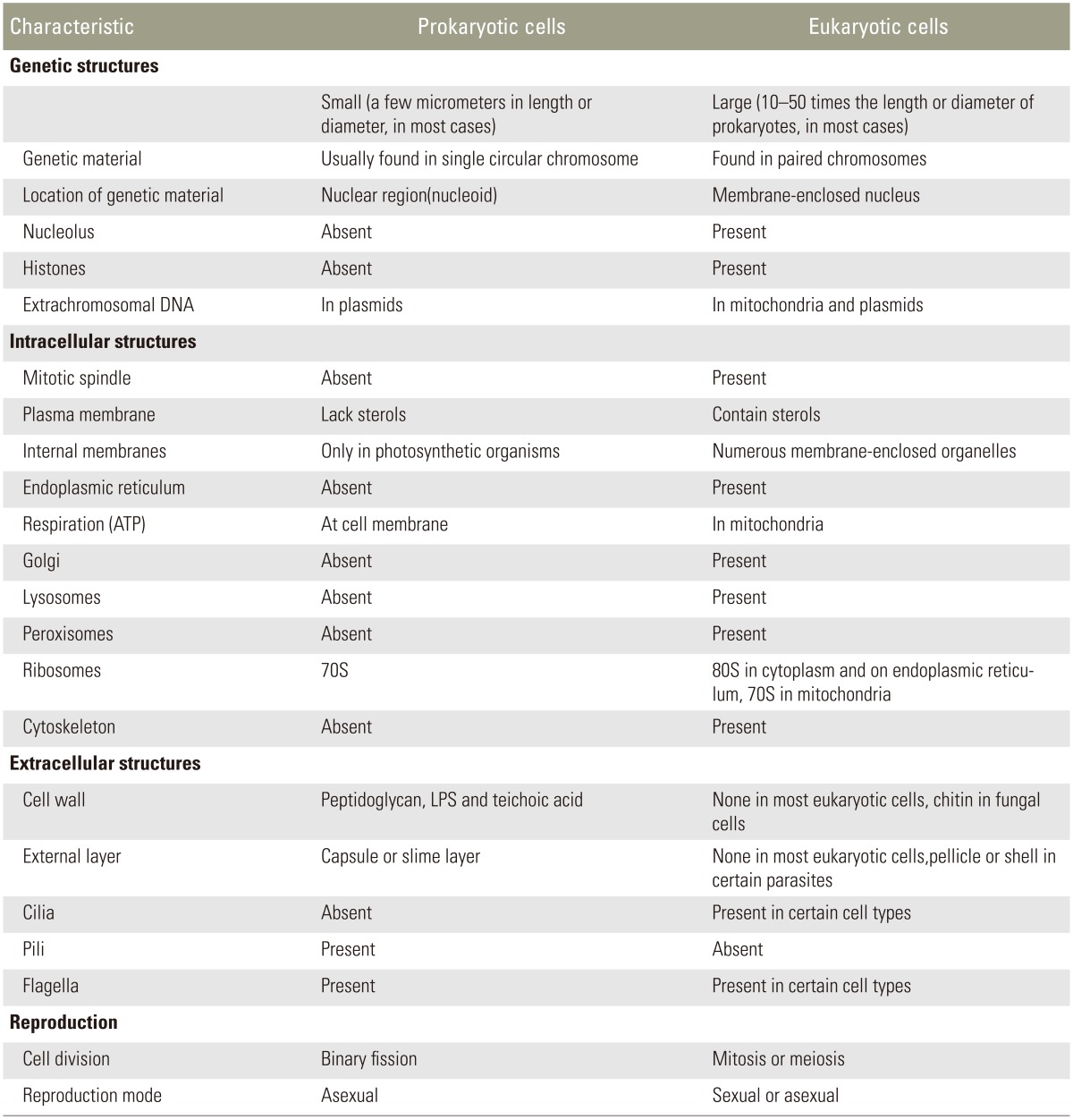
In describing the bacillary morphology and better defining the host-pathogen relationships, a summary of the structures of the host cell (eukaryotic cells) and bacterial pathogen (prokaryotic cells) is provided in Table 1. As Table 1 lists the major differences between the two cell types, an eukaryotic cell is bigger and far more complex than a prokaryotic one in almost every way [811]. All prokaryotes are single-celled organisms, having no membrane-enclosed nucleus and this includes bacteria. On the other hand, eukaryotes have a membrane-enclosed nucleus per cell, and this includes human cells.
M. tuberculosis is a tiny thin obligate aerobic (3 ┬Ąm in length and 0.5 ┬Ąm in width), acid-fast and a Gram-positive pathogen. As mentioned above, it can also be a facultative anaerobe. It is metabolically catalase-and phenylase-positive. It has no glycocalyx, pili or fimbriae on its cell wall surface for adhesion, and it is always present in planktonic form, even after repeated replications (Table 2). Even in a rich nutrient environment, its growth is slow and its size doesn't change. Tubercle bacilli do not display the classic ŌĆ£starvation response,ŌĆØ although other bacteria have it. Bacteria convert unsaturated fatty acids to cyclopropane during starvation and acid stress condition under which their membranes require stiffening, and cyclopropane conversion is an important factor in the pathogenesis of M. tuberculosis.
Tubercle bacilli are well-clad in the armor of a slippery thin waxy cell wall, and can survive poor nutrient milieu (famine) and oxygen deficiency (by becoming anaerobic). Tubercle bacilli's small size is not, however, the result of starvation.
Mycobacterial Behavior
Mycobacterial growth is inhibited within the necrotic environment by low oxygen tension and low pH. Its growth takes up to 6ŌĆō8 weeks for its colonies to become visible. The colonies clump together due to their hydrophobic lipid nature, and the bacillary generation time is 12 hours.
Oxygen is required for bacillary growth and has two roles against the bacilli being both aerobic and anaerobic. For aerobes, metabolically oxygen is essential for growth, though it can be facultative. However, many bacteria as anaerobes do not require oxygen for growth, and can actually die in presence of oxygen (obligate anaerobes). Thus, oxygen can also be detrimental to certain bacteria types.
Mycobacterial Cell Wall
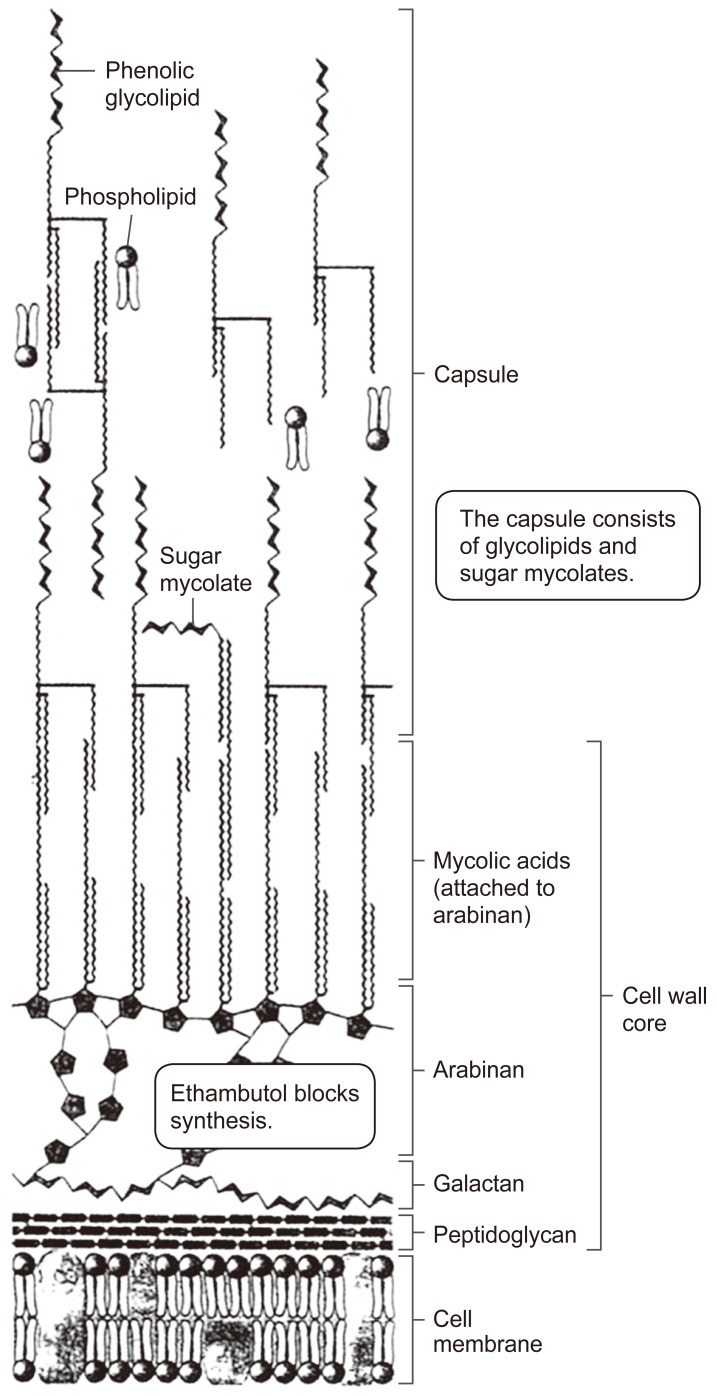
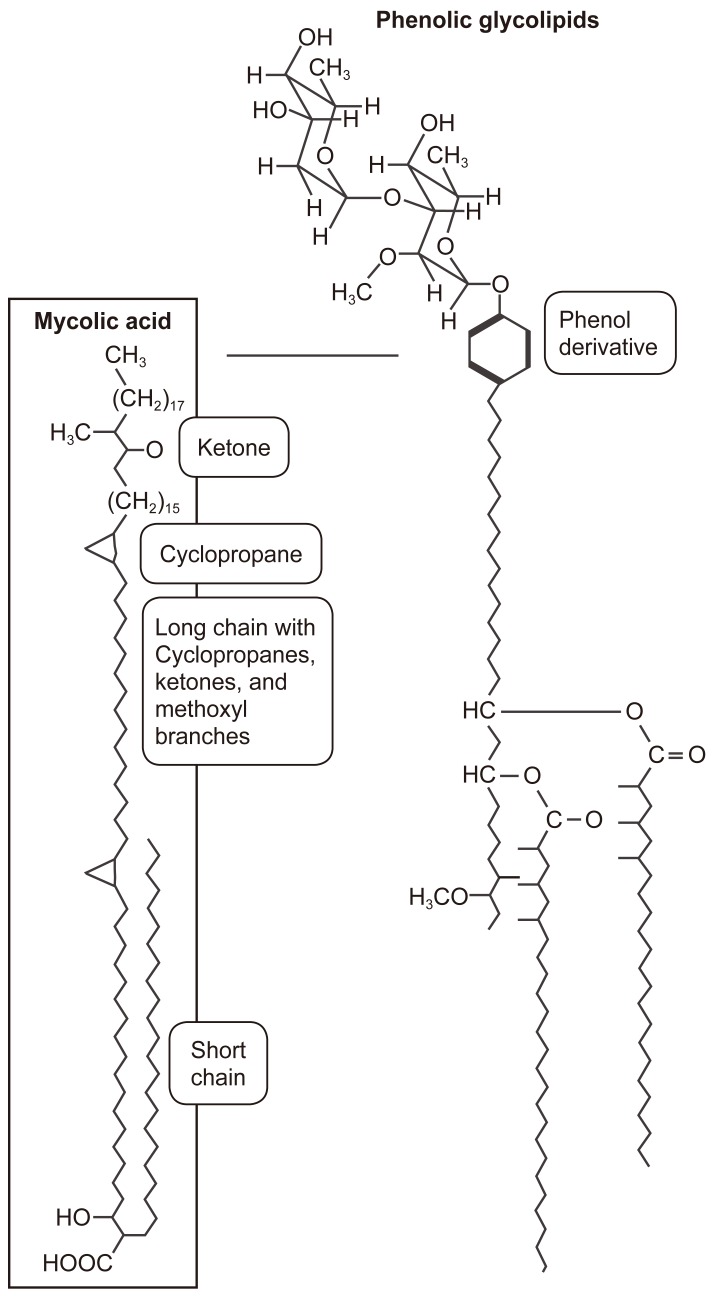
The cell wall in bacteria provides a protective structure, and for mycobacteria, the cell envelope includes unusual membrane lipids called mycolic acids, where there is a hydroxyl acid backbone with two hydrocarbon chains, one comparable in length to typical membrane lipids (of about 20 carbons), the other about three-fold larger. The longer chain includes ketones, methoxyl groups, and cyclopropane rings, and there are hundreds of different forms known (Figs. 1, 2).
The cell wall lipids in acid-fast organisms are involved in cell wall formation and they determine cell wall thickness, and affect virulence, protective and immune activities of the organisms (Fig. 3). The phenoloic glycolipids include a phenol group, also linked to sugar chains. The mycobacterial cell wall includes peptidoglycan linked to chains of galatose, called galactans. The galactans are attached to arabinans, polymers of the five-carbon sugar arabinose. Those arabinan-galactan polymers are known as arabinogalactans (Figs. 1, 2). The cell wall peptidoglycan is constructed inside the cell and transported to the outside, and it forms a meshwork for the cell wall and is also a target for a number of antibiotics. For example, arabinogalactan biosynthesis can be inhibited by ethambutol, a major antibiotic against tuberculosis.
The ends of the arabinan chains form ester links to mycolic acids, and the mycolic acids provide the basis for acid-fast staining in which cells retain the dye carbolfuchsin, an important diagnostic test for mycobacteria. In M. tuberculosis, the mycolic acids form a kind of bilayer interleaved with phenolic glycolipids. The extreme hydrophobicity of the phenol derivatives generates a waxy surface that prevents phagocytosis by macrophages. Mycolic acids in the cell envelope make up as much as 60% of the cell wall.
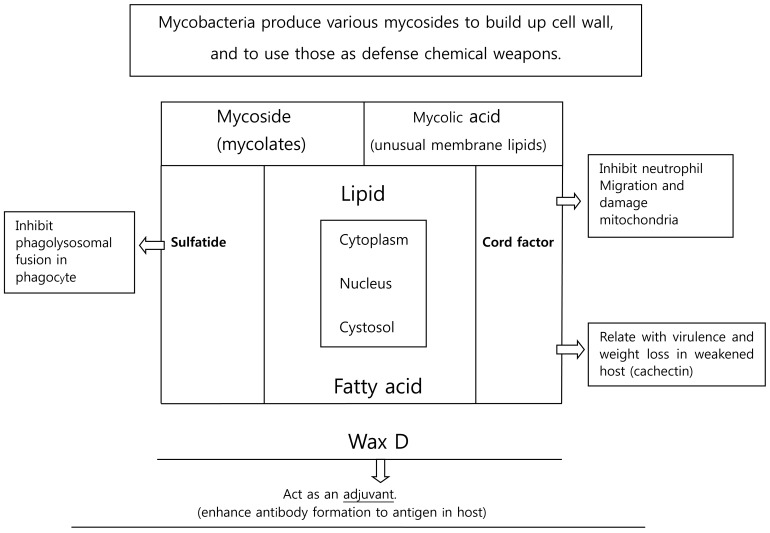
According to Cladwin and Tratter, mycolic acid can combine with other compounds to result in mycosides, cord factor, sulfatides, and wax D (Fig. 1):
(1) Mycoside (glycolipid) is formed by conjugation of mycolic acid with carbohydrates.
(2) Cord factor (trehalose) is formed by conjugation of two mycolic acid units with dissacharides. This factor is found only in the virulent strain of M. tuberculosis. In the host body, cord factor from M. tuberculosis leads to release of Tumor necrosis factor alpha (TNF╬▒) or cachectin, which in turn induces weight loss and cachexia.
(3) Sulfatides are the mycosides, which resemble the cord factor and formed by sulfate attached mycoside and dissacharide. It is known to inhibit the phagolysosomal fusion inside cell.
(4) Wax D is a complex mycoside, which has an adjuvant role and enhances antibody formation to an antigen, and activates the protective cellular immune system.
The M. tuberculosis chaperonin 10 (MtCpn10) is also known to associate with cell wall components, and it is believed to be a secreted protein related to drug resistance. The extreme hydrophobicity from the phenol derivatives of the mycobacterial cell wall generates a waxy surface that prevents macrophage's phagocytosis, and the waxy cell wall protects the organism against antiseptics, disinfectant and antibiotics. Given the significance of the cell wall to the organism, it may not be surprising that Mycobacterial behavior becomes somewhat different due to percentile differences in the cell wall chemical composition of each bacillus.
Living mycobacteria are more effective than the killed organism in eliciting protective immunity, and this difference is thought to be due to protective antigens, which are associated with only the growing microorganisms.
What Makes a Mycobacterium Pathogenic?
It is not easy for pathogens to produce disease in human, and virulence factors are required for a successful pathogen to persist in the host, as to cause disease, and escape host defense so that the infection can continue. In addition, virulence depends on various genetic factors. Justifiably, the topic of virulence factors has long been a focus of research for more effective therapies against various pathogens.
A pathogen must be able to have and accomplish the following:
(1) Potential pathogen must be able to adhere, penetrate, and persist in host cells (ŌĆ£get-in-and-stay-inŌĆØ rule), but tubercle bacilli cannot adhere and penetrate.
(2) A pathogen must be able to avoid, evade, or compromise the host defense mechanisms.
(3) A pathogen must damage the host tissue or organ, and permit the spread of the infection.
(4) A pathogen must be able to exist in one host and infect another host (transmission of disease). M. tuberculosis has all the above-listed condition to cause the disease and spread it.
Virulence of Tubercle Bacilli
Bacillary pathogenicity has long been an issue in combating the organism. Mycobacterium is a problematic pathogen for the host defense because of its unique cell wall. For pathogens, virulence defines the potential to cause disease and it refers to how harmful a given pathogen is to a host. There are cases where organisms become pathogenic, but are only mildly virulent.
Two criteria is defined in assessing the bacterial virulence: lethal dose 50% (LD50) and infection dose 50% (ID50). Pathogens that have lower LD50 and ID50 values are the most virulent.
Because it grows very slowly, the Mycobacterial virulence depends only on the infection dose. In this review, we put more emphasis on infection and infection establishment rates, and also the reactivation rates.
Bacillary virulence is defined by its activities:
(1) It can cause high pathogenicity (causes disease at a high rate).
(2) The pathogen may become highly contagious (disease spread is rapid).
(3) Displaying drug resistance (high bacillary survival rates even with targeted therapies).
Virulence and Drug Response
With regards to sensitivity to various drugs, there are three kinds of responders, and a slow or a non-responder to a drug might suggest drug resistance:
(1) Normal responder (effective).
(2) Slow responders (clinical symptoms improve slowly).
(3) Non-responder (no drug efficacy).
Out of several known mycolic acid compounds, the cord factor in the mycobacterial cell wall is only found in the virulent strains. In addition, M. tuberculosis chaperonin 10 has been strongly implicated as an important virulence factor during infection [16].
Mycobacterial Cell Wall Structure for Nutritional Supply and Bacillary Growth
The thick waxy envelope with poorly developed scarce porins (nutritional channels) that excludes many antibiotics and offers exceptional protection from host defenses, enabling the tuberculosis pathogen to colonize its host over long periods. However, the mycobacterial envelope also retards uptake of nutrient. As a result, M. tuberculosis grows extremely slowly and is a challenge to culture and grow in the laboratory [123].
Essential nutrients are those compounds a microbe cannot itself make, but must gather from its environment if the cell is to grow and divide, and microbial cells obtain all the essential nutrients for growth from their immediate environment. Consequently, when an environment becomes depleted of one or more essential nutrients, the microorganism stops growing. The growth factor in the natural habitat of M. tuberculosis (human host) is nicotinic acid (NAD) and alanine.
Bacteria and Oxygen
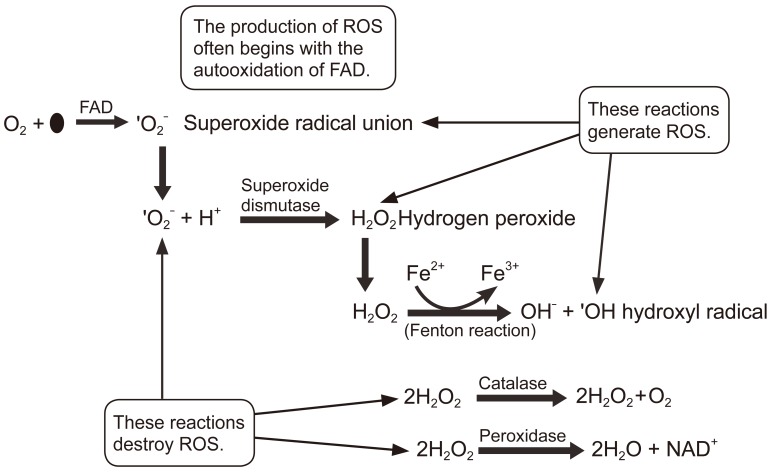
Oxygen and its relation to the living cells will be briefly discussed. Oxygen has both benefits and risks, depending on the organism and the cell type. For aerobes, oxygen is required for respiration and it is beneficial. Although oxygen is one of the most important requirements for bacterial growth, many bacteria not only do not require oxygen for growth, they can actually die in presence of ambient oxygen. Although we know that using oxygen as a final electron acceptor in aerobic metabolism maximizes the Adenosine triphosphate yield, oxygen also has a ŌĆ£dark sideŌĆØ as during normal bacterial respiration, a small portion of molecular oxygen can be in the form of superoxide free radicals (O2-). These free radicals are very unstable and capture electrons from other molecules, in a process that creates electron-deficient molecules, which can in turn also capture electrons, creating a cascade for generating radical molecules. The free radical formation and its damaging effects can eventually lead to the death of the bacterial cell.
Two types of bacteria can grow in presence of oxygen: aerobes, which are bacteria that require oxygen for growth, and facultative anaerobes, which can grow with or without oxygen. Both aerobic and facultative anaerobic bacteria produce an enzyme called superoxide dismutase (SOD) that can convert free-radical oxygen to molecular oxygen and peroxide (reaction below):
Unfortunately, the product H2O2 contains the peroxide anion O2-, which is just as toxic to the bacteria as free radical oxygen. This toxic peroxide anion is the active component of many antimicrobial agents such as hydrogen peroxide and benzole peroxide. To deal with this toxic anion, bacteria have developed two enzymes to neutralize it. The enzyme catalase converts hydrogen to water and oxygen.
The enzyme peroxidase converts hydrogen peroxide not to water plus molecular oxygen, but rather to just water:
Any organism that possesses NADH dehydrogenase 2 will in the presence of oxygen, inadvertently auto-oxidize the flavin adenine dinucleotide (FAD) cofactor within the enzyme and produce dangerous amount of superoxide radicals (*O2-). Superoxide will degrade to hydrogen peroxide, another reactive molecule. Catalase and peroxidase enzymes do not produce reactive oxygen species (ROS), but detoxify hydrogen peroxide. Iron is present as a cofactor in several enzymes, which can then catalyze a reaction with hydrogen peroxide to produce the highly toxic hydroxyl radical (*OH). All of these molecules can seriously damage DNA, RNA, proteins and lipids. As such, presence of oxygen creates an extreme environment in which survival requires special means of addressing the reactive free radicals generated (Fig. 4).
M. tuberculosis Defense Pathways
M. tuberculosis utilizes two pathways to defend itself for survival:
(1) Prevention of phagolysosomal fusion by the bacillary-produced sulfatide (a mycoside compound)
(2) By destroying the reactive oxygen species that are produced by phagocytes.
With the above means, the bacilli can defend themselves and remain unharmed. It should be noted that different bacterial species have evolved ways either to tolerate or avoid oxygen all together.
Both M. tuberculosis and phagocytes destroy excess remnant reactive oxygen species they produce with the aid of enzymes such as SOD, peroxidase and catalase (to remove H2O2). Aerobes also have repair enzyme systems that detect and repair macromolecules damaged by oxidation [123].
Phagocytosis is an effective non-specific immune response. However, it would be disastrous if white blood cells indiscriminately phagocytosed host as well as non-host structures. There are fail-safe mechanisms built into our immune defenses to prevent this from happening. Without these restraints, the human immune system would constantly attack itself, causing more problems than benefits. When one of these fail-safe mechanisms fails, autoimmune disease can arise.
Phagocyte's Recognition of Microbes and Alien Particles and Targeting Them for Elimination
7Recognition of bacteria and particle is the first step for phagocytosis to proceed. Macrophages and neutrophils must first recognize the surface of a microbe as foreign. When a phagocyte surface interacts with the surface of another cell, the phagocyte has to decide on whether the other cell is friend or foe, self or non-self. This self-recognition involves glycoproteins located on the white blood cell membrane, binding inhibitory glycoproteins present on all host cell membranes. The inhibitory glycoprotein on human cell is called CD47. Because invading bacteria lack these inhibitory surface molecules, they can readily be engulfed. M. tuberculosis is readily recognized and engulfed by phagocytosis [123].
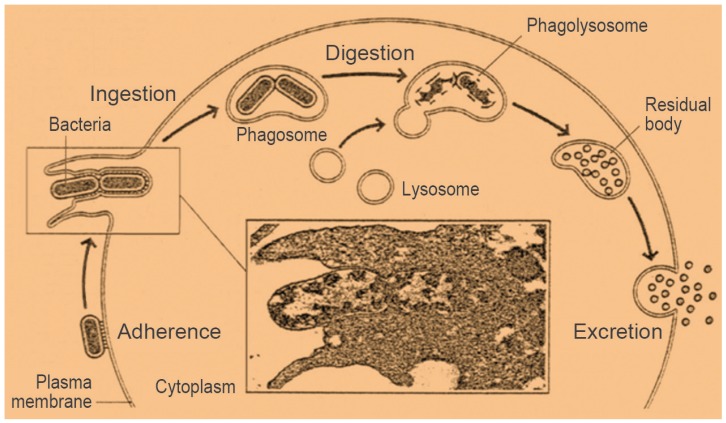
Organisms taken up into the phagocytic cells from their environment are internalized as large endocytic vesicles, called phagocytic vacuoles. These vesicles are also called ŌĆ£phagosomes.ŌĆØ These vacuoles fuse with lysosomes, forming large vesicles in which the ingested material is digested. This process is called ŌĆ£phagocytosis,ŌĆØ which is defensive rather than nutritional (Fig. 5). The phagocytic cells ingest and digest microorganisms that gain entry to the body. In addition, macrophages play a scavenging role, ingesting damaged cells and cellular debris from the circulating system [23].
Oxygen-Independent and -Dependent Killing Pathways for Phagocytes Against the Mycobacterium
During phagocytosis, the cytoplasmic membrane of the phagocyte causes a directed cell shape change, which engulfs the bacterium, thus producing an intracellular phagosome. Subsequently, phagosome-lysosome fusion (producing a phagolysosome) results in both oxygen-independent and oxygen-dependent killing pathways [123].
Oxygen-independent mechanism (the enzymatic pathway) include enzymes like lysozymes to breakdown bacterial cell wall, compounds such as lactoferrin to sequester iron away from the microbe, and defensins (small cationic antimicrobial peptides).
Oxygen-dependent mechanisms are probably activated through the toll-like receptors. Oxygen-dependent mechanisms kill through the production of various oxygen radicals. NADPH oxidase yields superoxide ion (*O2), hydrogen peroxide, and ultimately, hydroxyl radical (*OH) and hydroxyl ion (OH-).
Myeloperoxidase, present only in neutrophils, converts hydrogen peroxide and chloride ions to hypochlorous acid (HOCl), which is then involved in the digestion of the phagosome contents.
Macrophages, mast cells and neutrophils also generate reactive nitrogen intermediates that serve as potent cytotoxic agents. Nitric oxide (NO) is synthesized by NO synthetase. Further oxidation of NO by oxygen yields nitrate (NO2) and nitrate (NO3-) ions. These reactive oxygen species attack bacterial membranes and proteins. These mechanisms cause the large increase in oxygen consumption noted during phagocytosis, called the ŌĆ£oxidative burst.ŌĆØ The reactive chemical species formed during the oxidative burst does little to harm the phagocyte as the burst is limited to the phagosome and that the various reactive oxygen species such as superoxide are very short lived. Although these phagocytes are very good at clearing infection agents, many bacteria have developed ways to resist this aspect of innate immunity.
Tissue Inseminated Tubercle Bacillary Behavior Against Phagocytes
Both host phagocyte and mycobacterium survive independently as separate entities, though mycobacterium can possibly survive inside the macrophage, serving as a host. Interestingly both the host and parasite have very similar intracellular morphology and functions, they also have great structural differences with regard to the cell wall of the parasite (Figs. 1, 2).
Tubercle bacilli may in small numbers in a given lesion, but they are better defensively armed than the phagocytes. Tubercle bacilli also behave differently according to extra- and intra-cellular environmental changes such as pH and oxygen tension without having structural changes.
Tubercle bacilli are resistant to killing by macrophages, and can be found alive within such macrophages (intracellular mycobacteria). This resistance against host defense can lead to long-term (chronic) infection and formation of granulomas. This is despite the host mobilizing considerable cellular forces including phagocytes, producing various chemical weapons (chemokines and lysosomal enzymes including leukocidin) and having the immune system to fight against the mycobacterium.
Phagocytosis is selective for particles recognized as being foreign by the body. For the phagocytic killing processes against the mycobacterium, the first step is the phagosome formation through the process of entirely engulfing the bacilli by phagocyte-pseudopods, followed by entry into the phagocyte. The phagosome in the phagocyte cytoplasm then fuses with a lysosome by virtue of their similar membrane structures. This fusion allows the lysosomal enzymes to come into contact with the mycobacterium and destroy it. However, in tuberculosis, the phagolysosomal fusion process is hindered or inhibited by the mycobacterium-produced sulfatide. Thus, some of the engulfed mycobacteria eventually escape from a phagosome and enter the cytoplasm of macrophage where they can increase in numbers. With enough numbers outside of the phagocytes, the bacilli can spread lymph nodes where again they can enter the blood and distribute throughout the body.
Mycobacterial Survival in the Host Tissues
Mycobacteria survive inside granulomas, which are bodies made-up of host defense cells such as neutrophils, macrophages, T-cells, B-cells, dendrite cells, fibroblasts and matrix components. In addition, the activated macrophages aggregated in granulomas grow into gigantic cells. The question is how mycobacteria survive the initial contact with the phagocytic cells that are programmed to phagocytose and kill them! To fight against this host phagocytic process, mycobacterium produces sulfatides, cord factors and wax D, all components of the main cell wall. Cord factor inhibits neutrophil migration and damages mitochondria, and is also related to virulence of the pathogen and systemic weight loss in the host body (via TNF╬▒/cachectin production). Also, sulfatides inhibit phagolysosomal fusion [3].
Effect of M. tuberculosis-Produced Substances on the Host Body
Two substances are of note that can affect the host body: the first is cachectin (TNF╬▒, a compound induced by the mycoside-origin cord factor), which emaciates the patient, and the second is the chaperonin 10 (heat shock 10 KDa protein 1, Hsp 10) [131415] and is responsible for osteolytic activity and virulence. For example, recombinant M. tuberculosis cpn 10 is a potent stimulator of bone resorption in bone explant cultures, and induces osteoclastic recruitment, while inhibiting the proliferation of an osteoblast bone-forming cell line [1314]. However, it is clear that chaperonin 10 doesn't directly participate in the local bone destruction at the infection site.
Tissue and Organ Damage by the Tuberculosis Mycobacterium
Tuberculosis is a necrotizing infection and a granulomatous lesion is formed in the host body. The lesion is a circumscribed mass or nodule consisting mainly of histiocytes. Frequently, the center of the tubercle will have caseous necrosis together with tissue damage. In addition, bacterial pathogens can produce digestive enzymes and/or toxins, which can further damage the tissue. The tubercle bacilli, however, do not produce enzymatic osteolytic substances or toxins. Some of the infection-associated host tissue damage is due to overcompensation by the host defense, where the host defense reactions designed to fight the infection are too severe and damage the host tissue. The pathogen uses multiple methods to survive and thrive, while the host defense (the immune system) becomes involved immediately in an attempt to kill or expel the pathogens and protect its own tissue and organs from damage. The host defense for the invading pathogen is the key to protect the host body as the body mobilizes two immune systems to expel or kill the pathogen: innate and adaptive immune systems [2314].
Bone Destruction at the Tuberculous Lesion Site
It is assumed that the host bone tissue damage is caused primarily by tubercle formation and its necrosis, but also by the locally produced inflammatory substances such as PGE2 [79]. However, the participation of free oxygen radicals and localized osteoclast's activity in bone destruction at the tuberculous focus have not yet been fully elucidated.
Dekel and Francis [17] recommended the use of NSAIDs to inhibit bone resorption in acute osteomyelitis. PGE2, which is produced by the bacterial inflammatory organisms, not only induces osteolysis, but also leads to osteoid formation, which is concentration dependent. Based on this information, we (the senior author) recommended adding NSAIDs to the antituberculous chemotherapy regimen for a few weeks to inhibit inflammatory bone resorption [791012].
Mycobacterial Response to Implants
Various implants are already proven to be inert to the mycobacterium, as mycobacterium doesn't produce adhesion molecules known as biofilm (glycocalyx). Consequently, implants do not induce biomaterial-centered infection. Thus, various biomaterials for reconstruction of the destroyed bone and/or joint in tuberculosis can be safely used, and also they do not interfere with the effectiveness of chemotherapy, because all tubercle bacilli are present in a planktonic form and in a naked state. Thus, no impairment of drug passage through the porins is thought to occur.
Mycobacterial Response to Chemotherapy
It is most important to remind all the treating physicians that the thick waxy envelope of tubercle bacilli excludes many antibiotics, and offers exceptional protection from host defense, enabling bacilli to colonize their hosts over long periods. The mycobacterial envelope also retards uptake of nutrients. As a result, bacilli grow extremely slowly, and it is a challenge to culture them in the laboratory. Therefore, for the patient, 9ŌĆō12 months of the combined chemotherapy is needed [123].
The treating physician should make accurate diagnosis of osteoarticular tuberculosis at the earliest stage, and then start a multidrug shotgun chemotherapy at all potential targets and continue over 9ŌĆō12 months (ŌĆ£Fire early and hit everythingŌĆØ). Thus, tuberculosis can be medically aborted without sequellae and the development of complicated osteoarticular tuberculosis that can lead to surgical management.
Chemotherapy for a tuberculosis patient usually consists of a triple therapy involving isoniazid (INH), pyrazinamide (PZA) and rifampicin (RFP). These three drugs are taken once a day for two months, followed by a regimen of INH and RFP for nine more months. If the tubercle bacilli strain is drug resistant, the initial regimen should also include ethambutol.
It is good news that a new class of drugs, known as pyronins, which can replace rifampicin, has become available. When the mycobacterium becomes resistant to rifampicin, pyronins are available as also being able to target the RNA polymerase (RNAP), similar to refampicin
It may be concluded that tubercle bacilli in osteoarticular tuberculosis can be easily suppressed or eradicated by an orchestrated management between the patients and the physicians regardless of the bacillary chemosensitivity and/or chemo-resistance. Thus, it can be said that osteoarticular tuberculosis including spinal tuberculosis is a treatable medical condition.
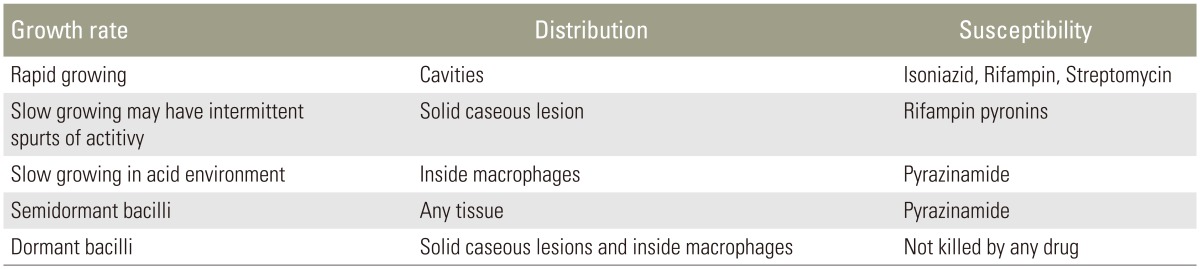
The effect of antituberculous agents depends on the physiological stage of the tubercle bacilli: whether in replicating and non-replicating stages. That is, at the given time, the treatment reflects the recognized mode of action of those antituberculous agents (Table 3).
Mechanisms of Action of Antituberculous Agents
1. Isoniazid (INH) is a bacteriostatic and narrow spectrum antibiotic. It becomes active only in presence of an enzyme, Kat G, produced by M.tuberculosis. It inhibits the synthesis of mycolic acid.
2. Streptomycin acts through its binding to the 30S ribosomal subunit, which affects polypeptide synthesis, and ultimately leads to inhibition of translation. Streptomycin is bacteriocidal and broad-spectrum.
3. Pyrazinamide is converted to bacteriocidal pyrazinoic acid inside the bacterial cell by pyrazinamidase/nicotinamidase. Pyrazinamide appears to kill a population of semidormant tubercle bacilli that are not affected by other antituberculous drugs.
4. Ethambutol is bacteriostatic. Single ethambutol is actually not very effective. Ethambutol in concert with isoniazid, inhibits the incorporation of mycolic acid (arabinogalactan biosynthesis) into the growing bacterial cell wall. Combination of isoniazid, ethambutol and rifampicin is now the treatment of choice for tuberculosis, and the administration of this combination of drugs will also lower the potential for development of drug resistance.
5. Rifampicin is bacteriostatic and broad spectrum antibiotic, and targets the RNA polymerase.
6. Rapidly growing tubercle bacilli in the cavities are sensitive to isoniazid (INH), rifampicin and streptomycin.
7. Slowly growing bacilli with intermittent spurts of activity in solid caseous lesions are sensitive to rifampicin. If the bacilli are resistant to rifampicin, new drugs targeting the RNA polymerase, called ŌĆ£pyronins,ŌĆØ can replace rifampicin.
8. Intracellular (inside macrophages) and slowly growing mycobacteria in an acid environment are sensitive to pyrazinamide.
9. Semidormant bacilli in any tissue are sensitive to pyrazinamide.
10. Dormant bacilli in solid caseous lesions and inside macrophages do not respond to any drug. Only revived bacilli from dormant state can respond to chemotherapeutic agents.
In summary spinal tuberculosis is primarily a medical condition, though it can become a surgical condition as a complication, and surgery is the last resort. Only more effective drugs can conquer tuberculosis and cure the patients. A surgical knife cannot cure the disease, and serves only as an adjuvant in managing complicated spinal tuberculosis. Surgeons must be wise enough to have a skillful hand, but must not be solely ŌĆ£supertechniciansŌĆØ or ŌĆ£technomaniacs.ŌĆØ
As a reminder, all treating physicians should have an understanding of the bacillary behavior, cell wall structure and drug response as the first key step in successful and curative management of spinal tuberculosis.